Smith Lemli Opitzi sündroom on kaasasündinud arenguhäire, mida iseloomustavad muude ilmingute hulgas ka iseloomulikud näojooned, intellekti- ja õpiraskused, käitumisprobleemid ja väike pea (mikrotsefaalia). Lisaks oluliste elundite väärarengutele, nagu neerud, süda, suguelundid ja sooletrakt, ilmnevad selle haigusega lastel autismi ja tähelepanupuudulikkuse hüperaktiivsuse häire (ADHD) omadused.Enamikul neist, kellel on haigus, on sulandunud teine ja kolmas varvas ning mõnel võivad olla lisasõrmed. See seisund on suhteliselt haruldane, seda mõjutab umbes üks iga 20 000–60 000 imiku kohta.
Seitsekümmend neli / Getty ImagesSümptomid
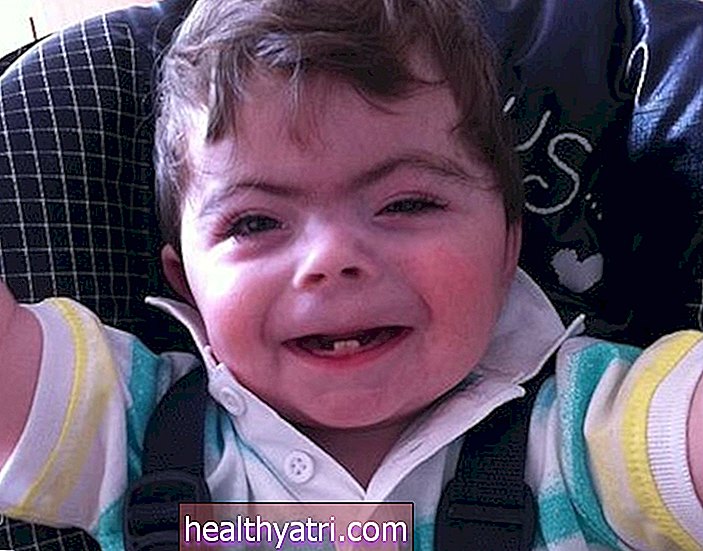
Smith Lemli Opitzi sündroomi tunnused ilmnevad sündides ja nende raskusaste on väga erinev. 80–99 protsendil nendest juhtumitest. neid omadusi on näha:
- Punutised varbad: haigusseisundi tavaliseks tunnuseks on teise ja kolmanda varba sulandumine, seisund, mida nimetatakse “sündaktüüliks”.
- Intellektuaalne puue: Ehkki selle määr võib varieeruda, viib see tingimus sageli õpiraskusteni.
- Ebanormaalselt väike kolju: Teine iseloomulik tunnus on keskmisest väiksem kolju suurus, seisund, mida nimetatakse mikrotsefaaliaks.
- Ebanormaalsed näojooned: Smith Lemli Opitzi sündroomiga inimestel on iseloomulikud näojooned, sealhulgas väiksem alalõug ja lai, lame nina. Harvadel juhtudel võivad inimestel olla rippuvad silmalaud, kassisilmad, väikesed või puuduvad silmad, samuti lai suu.
- Raskused toitmisel: imikutel võib see seisund põhjustada imetamisraskusi, mõjutades arengut.
- Madalam lihastoonus: sündroomi tavaliseks tunnuseks on keskmisest madalam lihastoonus.
On palju harvemaid sümptomeid, mis esinevad kõikjal 5–79 protsendil juhtudest, sealhulgas:
- Hammaste arengu kõrvalekalded: täiskasvanud hammaste varajane purse ja suurenenud igemed on Smith Lemli Opitzi sündroomi tunnused.
- Ebamäärased suguelundid: mõjutatud suguelundid võivad olla vähem määratletud. Mehed kogevad seda tõenäolisemalt, arenenud peenise ja laskumata munanditega.
- Tähelepanu puudulikkuse hüperaktiivsuse häire (ADHD): seda arenguhäiret iseloomustavad raskused käitumise ja impulsside reguleerimisel, samuti hüperaktiivsus.
- Autism: tuntud ka kui autismispektri häire (ASD), see seisund viib sotsiaalsete oskuste, kõne ja mitteverbaalse suhtlemise võimete ning korduva käitumiseni.
- Südame defektid: Smith Lemli Opitzi sündroomiga seotud südamepuudulikkuse hulka kuulub augu tekkimine seina kahe ülemise kambri vahel (kodade vaheseina defekt) või alumise kambri vahel (vatsakese vaheseina defekt).
- Muutunud käte anatoomia: Sellel haigusseisundil võivad olla eriti väikesed sõrmed ja varbad. Lisaks võib pöidla asend olla ebatüüpiline ka selle poolest, et see on randmele lähemal. Teatatud on ka riidest sõrmedest. On teatatud ka küüniste käest, sõrmede ebatüüpilisest kumerusest.
- Valgustundlikkus: paljudel juhtudel on kahjustatud inimeste nahk päikesevalguse suhtes eriti tundlik.
- Sage infektsioon: sündroomiga inimestel on kõrgem bakteriaalse infektsiooni oht.
- Lõhestatud keel: umbes viiel kuni 30 protsendil juhtudest on haigestunutel keeleline lõhk, mille ots on lõhestatud.
- Lülisamba kõrvalekalded: koos teiste selgroolülide deformatsioonidega võib haigusega kaasneda ka skolioos - selgroo külgmine kõverus -, samuti kyphosis või küürakas.
- Krambid: selle haigusega inimestel on krambihoogude tekkimise tõenäosus suurem.
- Tahtmatud silmaliigutused: sündroomiga võivad kaasneda ka kontrollimatud ja kiired silmaliigutused (nüstagm).
Põhjused
Smith Lemli Opitzi sündroom on geneetiline häire, mis on põhjustatud DHCR7 geeni mutatsioonist. See geen reguleerib olulist ensüümi 7-dehüdrokolesteroolreduktaasi, mis on seotud keha kolesterooli tootmisega. Kolesterool on oma funktsioonide hulgas rakumembraanide peamine komponent ja aitab moodustada müeliini - ainet, mis kaitseb ajurakke (neuroneid). See mängib olulist rolli ka korralikul seedimisel.
DHCR7 mutatsioon põhjustab 7-dehüdrokolesteroolreduktaasi puudust, põhjustades kolesterooli tootmise defitsiiti. Samuti võimaldab see kolesterooli toksilistel kõrvalproduktidel kehas kuhjuda, mis takistab arengut ja kasvu mitmes kehasüsteemis. Siiani uuritakse täpset mehhanismi, kuidas see kolesteroolipuudus põhjustab Smith Lemli Opitzi sündroomi.
Geneetiline defekt järgib seda seisundit nn autosomaalseks retsessiivseks mustriks, mis tähendab, et geeni mõlemad eksemplarid - üks mõlemalt vanemalt - on selle arenguks vajalikud. See tähendab, et haigusseisundi vanemad kannavad geeni, kuid neil pole tingimata ise sümptomeid.
Diagnoos
Nagu teiste kaasasündinud haiguste puhul, hõlmab Smith Lemli Opitzi diagnoosimine nii füüsiliste sümptomite hindamist kui ka 7-dehüdrokolesteroolreduktaasi ja kolesterooli suhte testimist. Seda tehakse kahtlustatavate juhtumite vereanalüüside abil. Lisaks võib sünnieelne geneetiline testimine tuvastada ka DHCR7 geeni mutatsioone, mis põhjustavad seisundi arengut.
Ravi
Selle tingimuse võtmine hõlmab koordineeritud jõupingutusi; kuna sellele haigusele pole otsest ravi, tuleb sümptomeid ja ilminguid tõhusalt juhtida. Sellised lähenemisviisid hõlmavad järgmist:
- Kolesterooli lisamine: Kuigi selle lähenemisviisi tõhususe hindamiseks on vaja rohkem uuringuid, võib kolesteroolirikas dieet - lisaks toidulisandite võtmisele - aidata mõningaid sümptomeid vähendada.
- Füsioteraapia: Füüsilise ja tööteraapia lähenemisviisid, kui need on õigeaegselt läbi viidud, võivad aidata selle haigusega seotud puudega inimestel.
- Meditsiiniline ravi: Smith Lemli Opitzi sündroomi mõningate füüsiliste sümptomite, sealhulgas seedimisraskused, nägemisprobleemid, samuti näo ja muud deformatsioonid, on olemas.
- Järelevalve: selle seisundi edukas juhtimine nõuab füüsiliste sümptomite, arengupeetuste ja toitumistegurite järjepidevat jälgimist.
Prognoos
Hea uudis on see, et kui Smith Lemli Opitzi sündroomi korralikult ravitakse ja piisavat arstiabi osutatakse, on selle haigusega inimestel eeldatav keskmine eluiga. See tähendab, et iseseisev elu on ebatõenäoline raske vaimupuude tõttu, mis sageli kaasneb selle sündroomiga. Eelkõige on tõsiste sümptomitega imikute ellujäämine tõsiselt mõjutatud ja paari kuu jooksul on võimalik surm.
Toimetulek
Suurem kaasasündinud häire, nagu Smith Lemli Opitzi sündroom, kujutab endast olulist väljakutset nii mõjutatud isikule, nende perele kui ka arstidele. Ehkki edukas juhtimine on võimalik, pole kahtlust, et sellest koormast on märkimisväärne psühholoogiline langus. Neile, kes on selle haigusega kellegi eest hoolitsemisele seatud, võivad olla abiks nõustamis- või puuetega inimeste abirühmad. Nimelt kogub selliseid ressursse nagu lingid uusimatele uurimis- ja tugiteenustele Smith Lemli Opitzi / RSH fond.
Sõna Verywellist
Seisund, mis on nii kurnav ja keeruline, mis võib mõjutada nii paljusid elukvaliteedi aspekte, võib tunduda valdav. See tähendab, et Smith Lemli Opitzi sündroomi ravimeetodeid ei täiustata ja täiustata pidevalt, vaid selle häire uurimine jätkub. Kui meditsiinikogukond saab rohkem teada selle seisundi põhjustest ja tagajärgedest - samuti ravimeetodite tõhususest -, paranevad mõjutatud inimeste prognoosid ja elukvaliteet ainult.


.jpg)